Data loading#
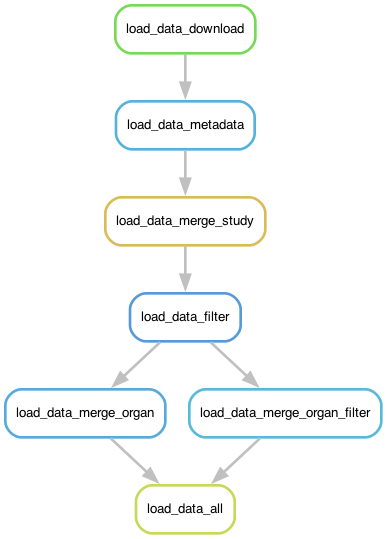
Workflow#
Given a TSV file and a schema mapping, the pipeline does the following:
Examples of configuration files are: test/config.yaml and test/datasets.tsv.
By default, dataset configurations are available under configs at the top-level pipeline (git root of this repository), but they can be modified or replaced by custom files.
Load data#
Files can either be read from a specified input file or downloaded from an URL, CELLxGENE or DCP directly. Which way a dataset is loaded, depends on the dataset mapping.
Aggregate Metadata#
The metadata rule adds additional dataset-level information that is included from the input TSV file.
This steps expects the data to follow
the CELLxGENE schema 3.0.0
and is extended as described below.
Other steps include:
adding external annotations if annotation file and columns are available in the TSV
saving donor IDs under
.obs['donor']inferring sample ID from input TSV and saving it under
.obs['sample']
The input AnnData objects must contain:
.Xraw counts, sparse format.obscolumns from the schema defined in the schema.obs.indexcell barcode
The output AnnData objects will contain:
.Xraw counts, sparse format.uns['meta']metadata from TSV file.obscolumns from CELLxGENE schema 3.0.0 and a subset of information in.uns['meta']fromEXTRA_COLUMNSfromscripts/utils.py.obs['dataset'],.uns['dataset']name of task/dataset.obs['organ'],.uns['organ']organ.obs['donor']donor ID.obs['sample']sample ID (inferred from input TSV).obs['barcode']cell barcodes as declared in index.obs['author_annotation']author annotation under theauthor_annotationcolumn of the input TSV.vargene information as specified in CELLxGENE schema 3.0.0.obs.indexunique cell identifiers e.g. dataset + numerical index
The AnnData is saved as a zarr file for a better speed to compression tradeoff compared ot gzipped h5ad files.
Merge Data#
This operation is applied by the following rules
merge_study: merge all datasets of a study if multiple datasets are available, else create a symlinkmerge_organ: merge all studies that belong to an organmerge_organ_filter: merge all cells removed by filtering per organmerge_subset: merge datasets by subset defined in the input TSV undersubset(overlapping subsets allowed)
The AnnData must contain all the slots described in Aggregate Metadata apart from the .uns slot.
Filter#
Filter cells per study depending on the config.yaml specification.
Two keys are available for controlling the filtering behaviour, filter_per_organ specifies global filter paramters
for all datasets per organ and filter_per_study allows for study specific filter options.
An example of an organ-level filter specification is shown below:
filter_per_organ:
blood:
cells_per_sample:
min: 50
max: 10000
mito_pct: 30
remove_by_colum:
dataset:
- Lee2020_2
All organ-level filtering decisions are applied per study.
The remove_by_column key can include any columns that are available in the anndata objects per study.
An example of per study filters shows that only the studies that require further filtering need to be overwritten. The filter options are the same as for the organ-level filters.
filter_per_study:
SchulteSchrepping2020:
remove_by_colum:
sample:
- Schulte-Schrepping_C2P01H_d0
- Schulte-Schrepping_C2P05F_d0
- Schulte-Schrepping_C2P07H_d0
- Schulte-Schrepping_C2P10H_d0
- Schulte-Schrepping_C2P13F_d0
- Schulte-Schrepping_C2P15H_d0
- Schulte-Schrepping_C2P16H_d0
- Schulte-Schrepping_C2P19H_d0
donor:
- C19-CB-0008
disease:
- influenza
Both keys can be empty or missing from the config file. In that case, no filtering is applied.
Preparing the input data#
In order to use the data loader module, you need to define the following files with the file locations and dataset-level metadata.
Dataset definiton file (
configs/datasets.tsv)Schema mapping (e.g.
configs/schema_mapping.tsv)Configuration file (e.g.
configs/imported/config.yaml)DCP metadata (optional)
Additionally, you need to prepare your input AnnData files to contain the metadata that is defined in your dataset definition file.
Dataset definition file#
The dataset definition file should specify which datasets you want to include for your analyses together with any additional dataset-level metadata.
Column |
Description |
|---|---|
dataset |
Name of the dataset, multiple datasets make up a study |
study |
Name of the study, used for aggregating datasets to study level |
organ |
Name of the organ, can be tissue or any other name for aggregating the atlas |
donor_column |
Column with donor IDs a donor is an individual who provided the sample |
sample_column |
Column with sample IDs ideally a sample is a subset of cells associated with an individual. The data should be deconvoluted, i. e., a sample mustn’t contain multiple individuals |
author_annotation |
Column with author annotations (needed for annotation quality assessment and label harmonisation) |
cell_type |
Optional. Column with cell ontology labels (needed to for different versions of the CELLxGENE schema, TODO: deprecate). If cell_type is missing from the file, it will be generated from author_annotation |
schema |
Name of schema to be mapped to |
url |
URL or path to the |
collection_id |
Only for |
dataset_id |
Only for |
project_uuid |
Only for |
annotation_file |
Optional. Any additional annotations that are not in the |
barcode_column |
Optional. Column in |
All other columns are optional and will be added to AnnData.uns['meta'].
Schema Mapping#
The data loader ensures that the data adheres to the CELLxGENE schema 3.0.0 specifications.
For datasets that do not adhere to that schema, the schema mapping file allows to provide a mapping of custom AnnData.obs columns to the ones defined in CELLxGENE.
Below is an example of a schema mapping for the schemas custom and dcp to cellxgene.
The column names are used to map the dataset to its corresponding schema.
cellxgene custom dcp
study study_PI project.contributors.name
sample sample_ID specimen_from_organism.biomaterial_core.biomaterial_id
donor_id subject_ID donor_organism.biomaterial_core.biomaterial_id
development_stage subject_developmental_state donor_organism.development_stage.text
sex sex donor_organism.sex
self_reported_ethnicity ethnicity_free_text donor_organism.human_specific.ethnicity.text
suspension_type biological_unit library_preparation_protocol.nucleic_acid_source
assay library_platform library_preparation_protocol.library_construction_method.text
organism species donor_organism.genus_species.text
cell_type cell_type
... ... ...
DCP metadata (optional)#
This file is optional and used for datasets for which users want to map additional HCA DCP metadata annotations.
The mapping should contain a study and a filename column, where filename is a TSV file that follows the DCP metadata schema.
The mapping does not have to include all the studies that you want to include in your analysis.
TODO: extend to other metadata input.
Configuration file#
The config.yaml file is the main configuration file of the pipeline and is included in the top-level workflow by default.
For data loading, you just need to define the location of the files defined above.
By default, configs/imported/config.yaml should already contain the correct paths to the previously described input files.
If you configured your datasets with different files than listed below, you need to update them accordingly.
dataset_meta: configs/datasets.tsv
schema_file: data/input/schema_mapping.tsv
dcp_metadata: configs/dcp_metadata.tsv
filter_per_organ:
...
filter_per_study:
...
For more information on Snakemake configuration files, please refer to the documentation.
Testing#
The test configuration and command are under `tests/’. All paths in the following are relative to the module root directory.
Prepare test data#
Before running the test pipeline for the first time, you must download a test dataset.
The following script downloads the “SchulteSchrepping” dataset and then copies it to the location that is defined in dataset.tsv.
bash test/download_test_data.sh -c1
This needs to be done only once.
Run pipeline on test configuration#
Activate the snakemake environment and call test/run_test_*.sh with run specific Snakemake parameters.
conda activate snakemake
bash test/run_test_cellxgene.sh -n
bash test/run_test_cellxgene.sh -c